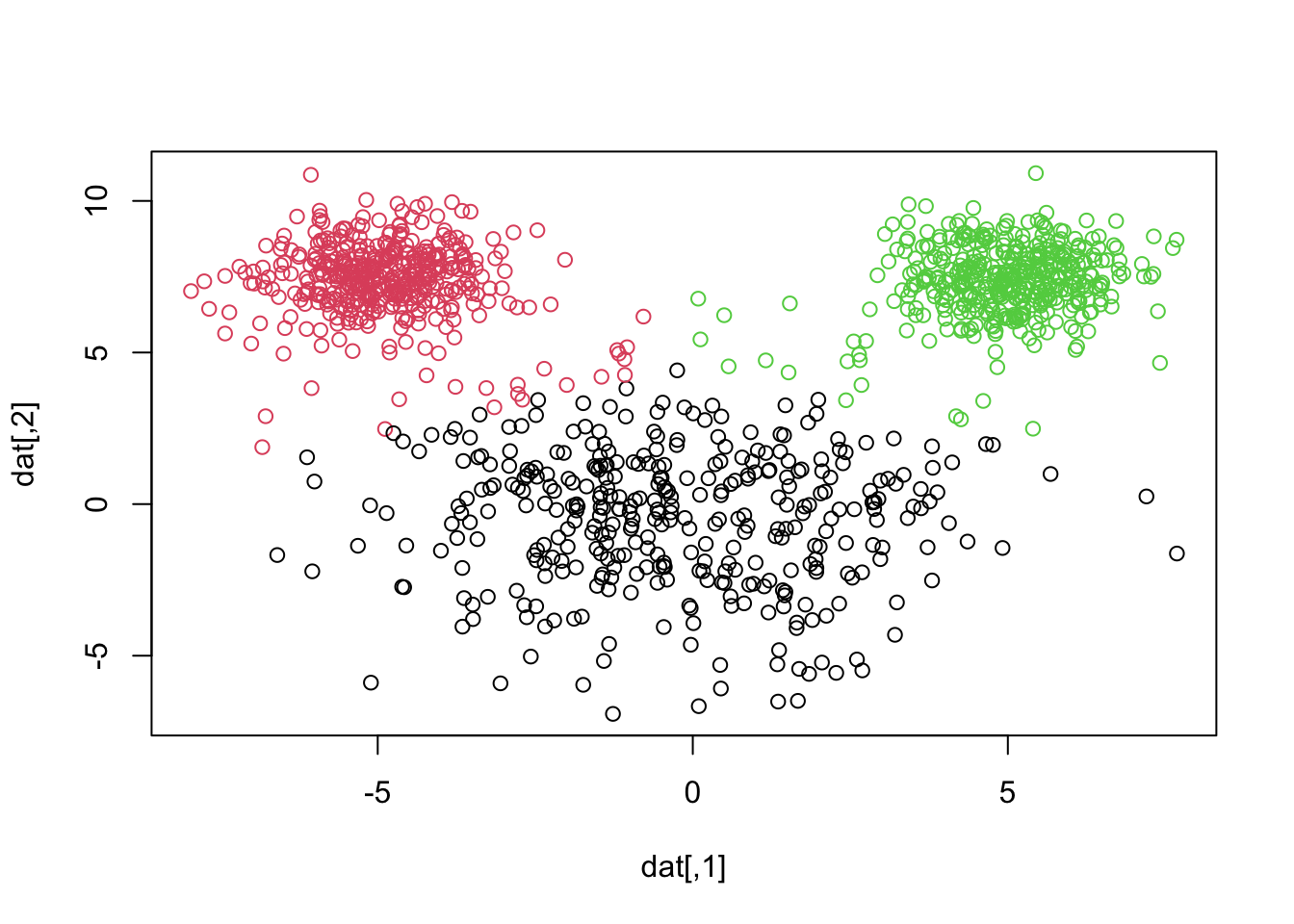
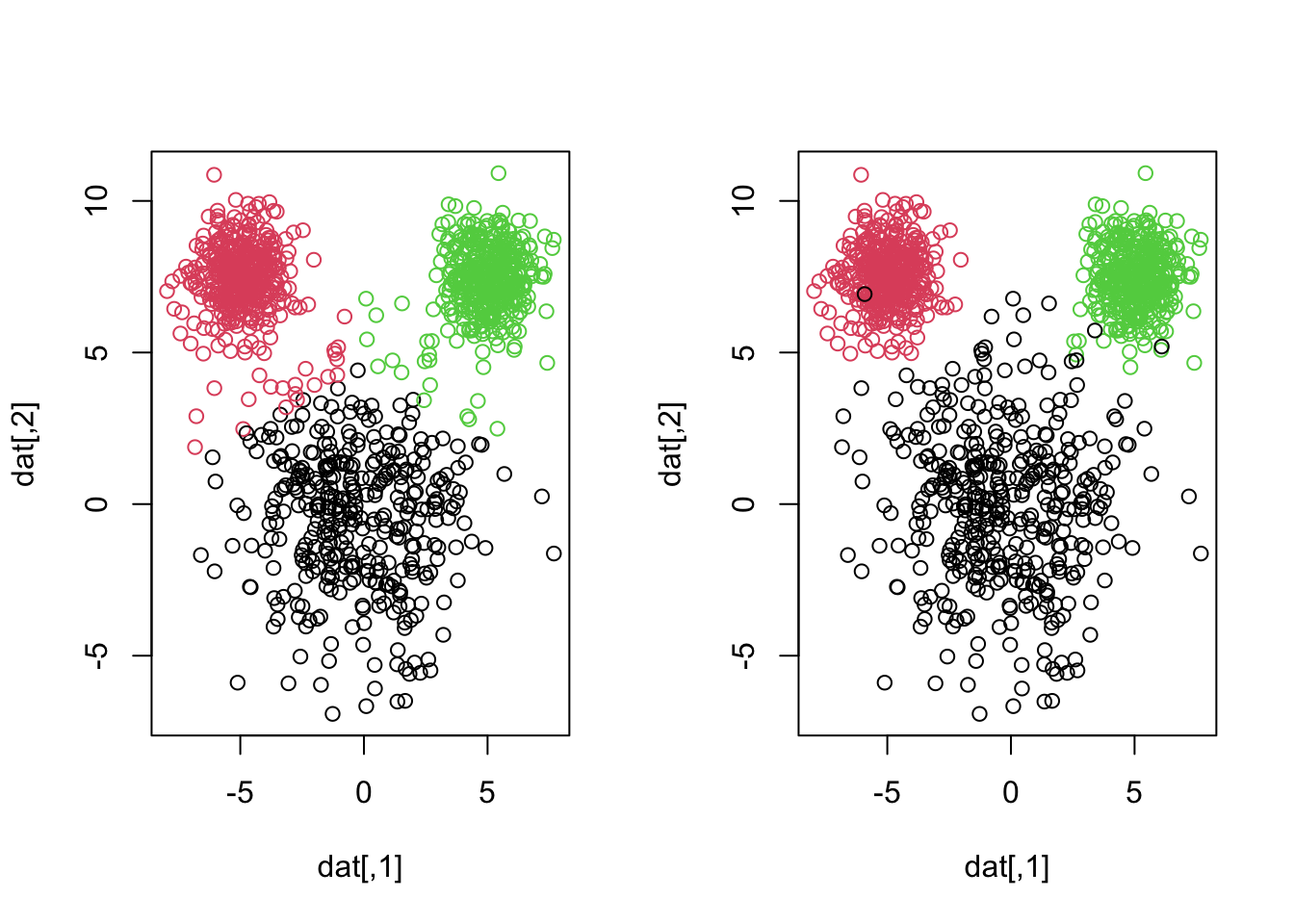
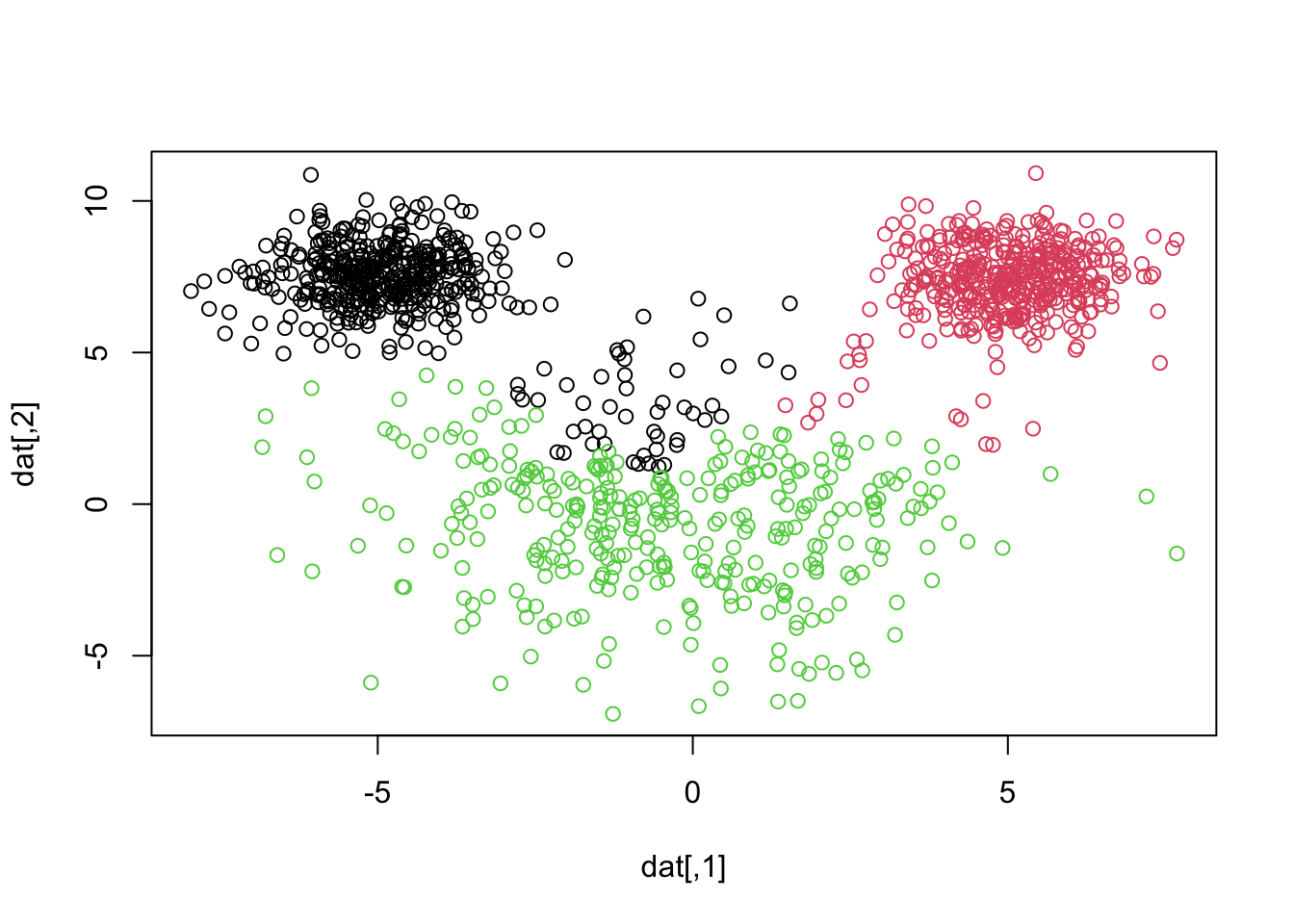
library(MASS)
set.seed(3173)
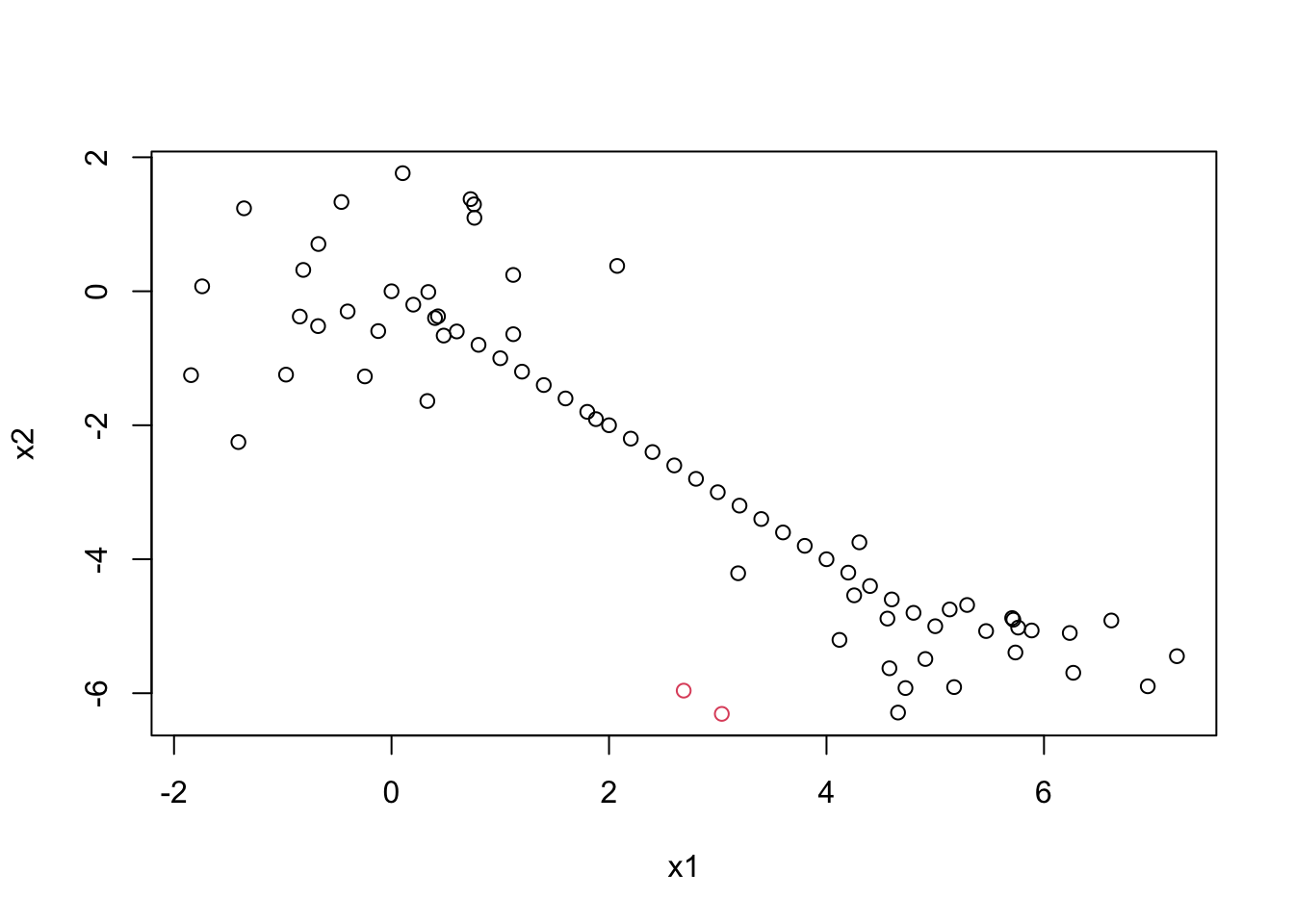
datagen1 <- mvrnorm(25, c(0,0), matrix(c(1,0,0,1),2,2))
datagen2 <- mvrnorm(25, c(5,-5), matrix(c(1,0,0,1),2,2))
datagen <- rbind(datagen1, datagen2)
x1 <- seq(from=0, to=5, by=0.2)
x2 <- seq(from=0, to=-5, by=-0.2)
newvalx <- cbind(x1,x2)
datagen <- rbind(datagen,newvalx)
plot(datagen)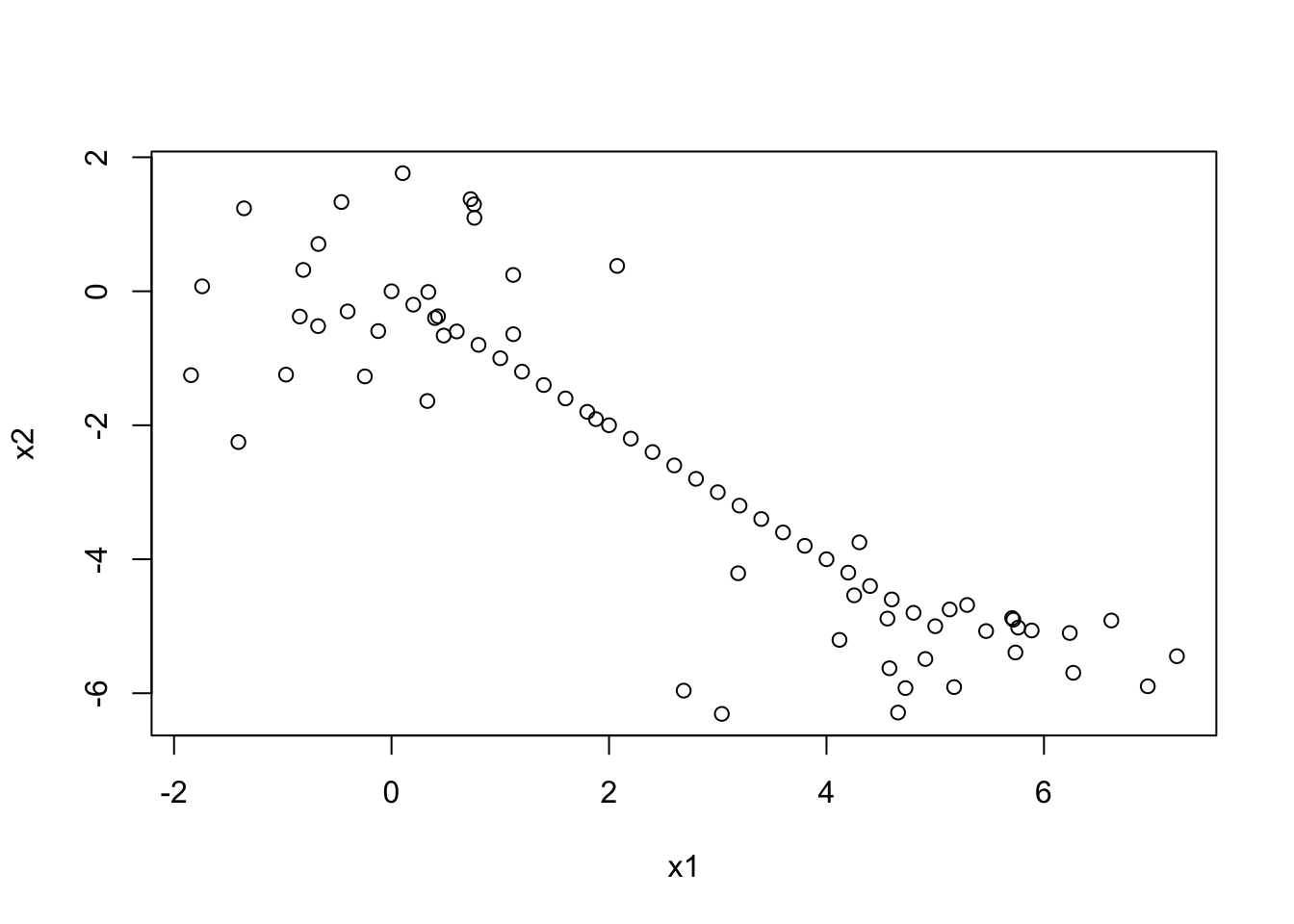
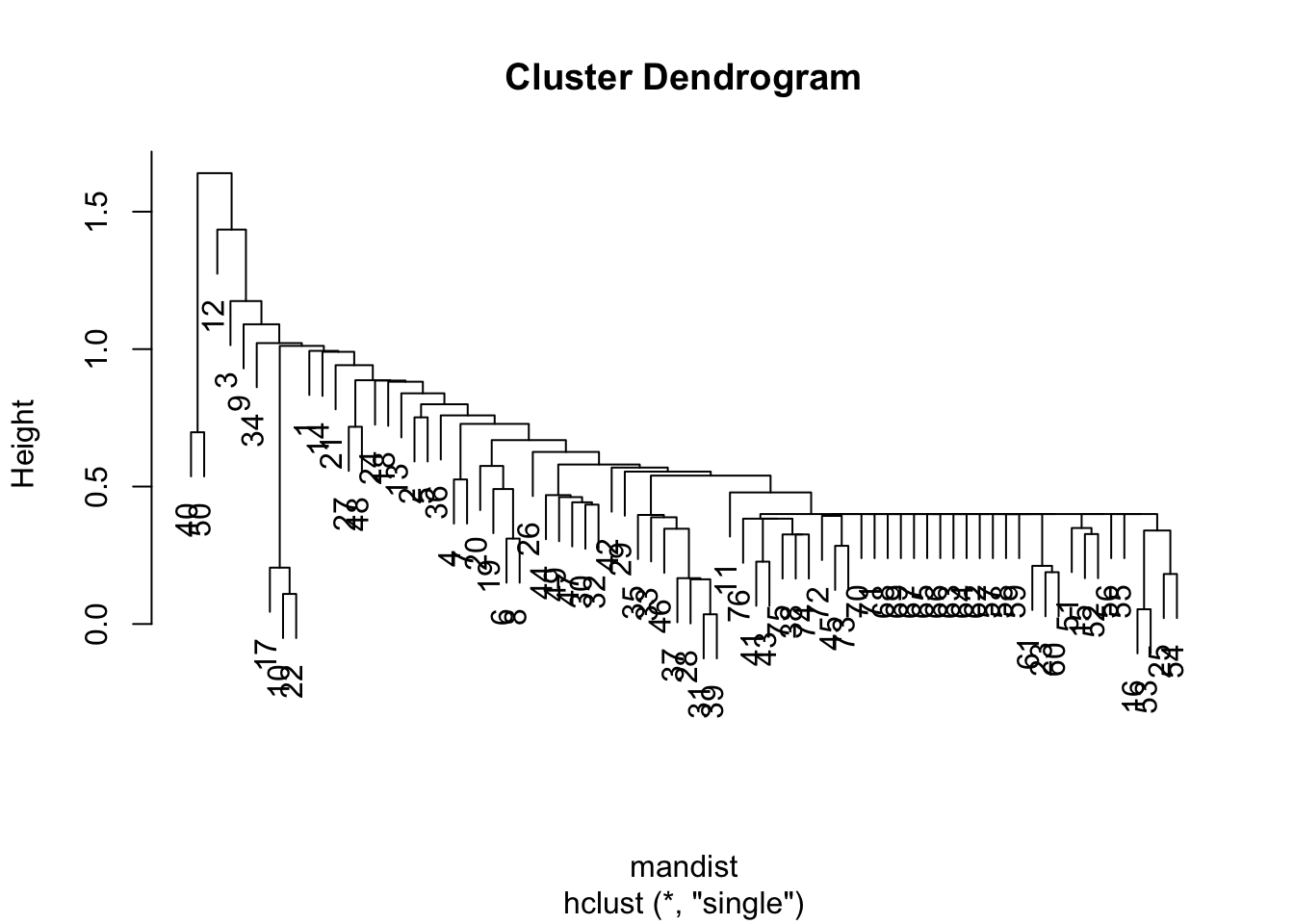
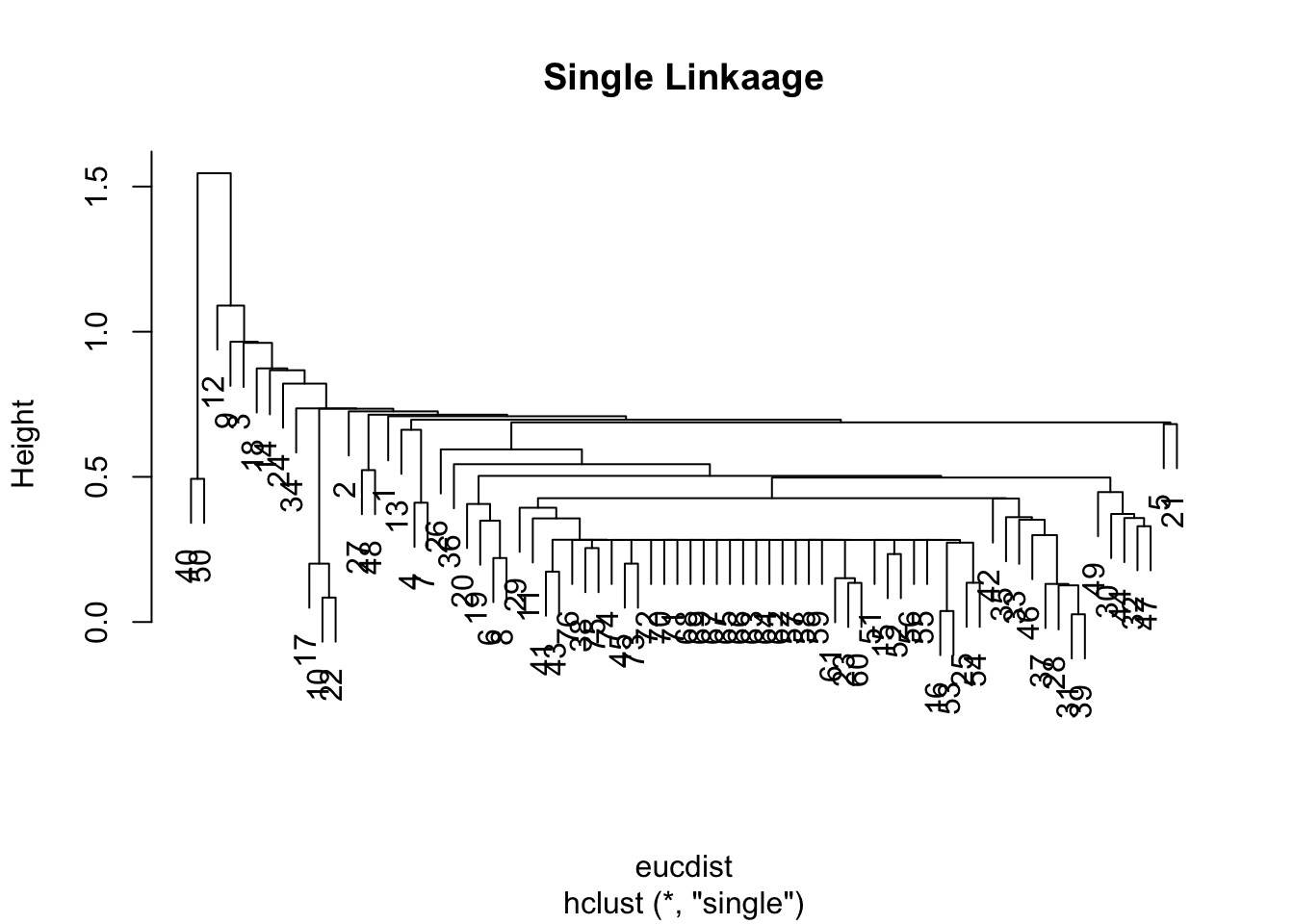
mandist <- dist(datagen,method="manhattan")
clus1 <- hclust(mandist, method="single")
plot(clus1)